 PDF(27228 KB)
PDF(27228 KB)

Bruton's tyrosine kinase knockout in macrophages attenuates diabetic kidney disease in the streptozotocin-induced mice
Zheng Zhichao, Fan Zhe, Wu Yonggui
PDF(27228 KB)
Bruton's tyrosine kinase knockout in macrophages attenuates diabetic kidney disease in the streptozotocin-induced mice
Objective To investigate whether Bruton's tyrosine kinase knockout (Btk-/-) in macrophages attenuates diabetic kidney disease in the streptozotocin (STZ)-induced mice. Methods Macrophages-specific Btk-/- mice and control mice (C57BL/6N) were randomly divided into WT group, diabetic group, Btk-/- group and Btk-/- diabetic group. The diabetic models were induced by STZ (50 mg/kg). After 12 weeks, relevant biochemical parameters and the histological changes of kidneys were detected. The expression of macrophages marker CD68 were detected by immunofluorescence, and the immunohistochemistry was employed to detect the expression of WT1 and Nephrin on renal podocytes. In addition, the expression of fibronectin (FN), collagen type IV (IV-Col), transforming growth factor-β1 (TGF-β1), iNOS, phospho (p)-Btk, interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), MAPK and NF-κB signaling pathway were detected by Western blotting. RT-PCR was used to detect the mRNA of IL-1β, TNF-α and monocyte chemotactic protein-1 (MCP-1). Results Compared with diabetic group, the mice in Btk-/- diabetic group had reduced albuminuria and attenuated kidney histopathology significantly, significantly increased WT1 and Nephrin, significantly decreased expression of CD68, FN, IV-Col and TGF-β1, and these changes were correlated with decreased of renal inflammatory cytokines such as IL-1β, TNF-α, MCP-1 and down-regulating MAPK and NF-κB signaling pathway (all P<0.05). Conclusion Macrophages-specific Btk-/- may protect the kidney of diabetic mice by reducing the expression of renal inflammatory cytokines in MAPK and NF-κB signaling pathway.
Diabetic nephropathies / Inflammation / Macrophages / Bruton's tyrosine kinase {{custom_keyword}} /
| [1] |
Immune modulation is now known to contribute to the development of glomerulosclerosis, tubulointerstitial fibrosis and end-stage renal disease in a large number of kidney diseases. Similarly, diabetic nephropathy is increasingly considered an inflammatory disease, with immune modulation being involved in both the development and progression of the disease. Infiltration of immune cells including macrophages, T cells, B cells and mast cells into the kidney has been reported. A number of pro-inflammatory cytokines and chemokines also play a major role in pathogenesis of diabetic nephropathy. Consequently, a variety of therapeutic strategies involving modulation of the immune response are currently being investigated in diabetic kidney disease. Copyright © 2013 Elsevier Ltd. All rights reserved.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [2] |
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [3] |
Evidence from renal biopsies has shown that macrophage accumulation in diabetic kidneys predicts declining renal function, suggesting a pathogenic role for these cells in diabetic nephropathy. Further evidence from animal models has shown that macrophages are the major immune cells infiltrating the kidney in type 1 and type 2 diabetes, and that they contribute to the development of renal injury and sclerosis. This review examines macrophages in human and experimental diabetic nephropathy, exploring the mechanisms of macrophage recruitment and activation, and the process of macrophage-mediated injury in diabetic kidneys. The ability of current therapies and novel anti-inflammatory treatments to reduce macrophage-mediated injury in diabetic kidneys also is considered, which has important implications for the future management of patients with diabetic nephropathy.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [4] |
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [5] |
Bruton's agammaglobulinemia tyrosine kinase (Btk) is a cytoplasmic tyrosine kinase important in B-lymphocyte development, differentiation, and signaling. Btk is a member of the Tec family of kinases. Mutations in the Btk gene lead to X-linked agammaglobulinemia (XLA) in humans and X-linked immunodeficiency (Xid) in mice. Activation of Btk triggers a cascade of signaling events that culminates in the generation of calcium mobilization and fluxes, cytoskeletal rearrangements, and transcriptional regulation involving nuclear factor-kappaB (NF-kappaB) and nuclear factor of activated T cells (NFAT). In B cells, NF-kappaB was shown to bind to the Btk promoter and induce transcription, whereas the B-cell receptor-dependent NF-kappaB signaling pathway requires functional Btk. Moreover, Btk activation is tightly regulated by a plethora of other signaling proteins including protein kinase C (PKC), Sab/SH3BP5, and caveolin-1. For example, the prolyl isomerase Pin1 negatively regulates Btk by decreasing tyrosine phosphorylation and steady state levels of Btk. It is intriguing that PKC and Pin1, both of which are negative regulators, bind to the pleckstrin homology domain of Btk. To this end, we describe here novel mutations in the pleckstrin homology domain investigated for their transforming capacity. In particular, we show that the mutant D43R behaves similar to E41K, already known to possess such activity.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [6] |
BTK and ITK are cytoplasmic tyrosine kinases of crucial importance for B and T cell development, with loss-of-function mutations causing X-linked agammaglobulinemia and susceptibility to severe, frequently lethal, Epstein-Barr virus infection, respectively. Over the last few years, considerable efforts have been made in order to develop small-molecule inhibitors for these kinases to treat lymphocyte malignancies, autoimmunity or allergy/hypersensitivity. The rationale is that even if complete lack of BTK or ITK during development causes severe immunodeficiency, inactivation after birth may result in a less severe phenotype. Moreover, therapy can be transient or only partially block the activity of BTK or ITK. Furthermore, a drug-induced B cell deficiency is treatable by gamma globulin substitution therapy. The newly developed BTK inhibitor PCI-32765, recently renamed Ibrutinib, has already entered several clinical trials for various forms of non-Hodgkin lymphoma as well as for multiple myeloma. Experimental animal studies have demonstrated highly promising treatment effects also in autoimmunity. ITK inhibitors are still under the early developmental phase, but it can be expected that such drugs will also become very useful. In this study, we present BTK and ITK with their signalling pathways and review the development of the corresponding inhibitors. © 2013 John Wiley & Sons Ltd.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [7] |
In this study we have identified members of the Toll-like receptor (TLR) family (namely, TLRs 4, 6, 8, and 9) as proteins to which the intracellular protein tyrosine kinase, Bruton's tyrosine kinase (Btk), binds. Detailed analysis of the interaction between Btk and TLR8 demonstrates that the presence of both Box 2 and 3 motifs in the Toll/interleukin-1 receptor domain was required for the interaction. Furthermore, co-immunoprecipitation experiments revealed that Btk can also interact with key proteins involved in TLR4 signal transduction, namely, MyD88, Mal (MyD88 adapter-like protein), and interleukin-1 receptor-associated kinase-1, but not TRAF-6. The ability of Btk to interact with TLR4 and Mal suggests a role for Btk in lipopolysaccharide (LPS) signal transduction. Stimulation of the human monocytic cell line THP-1 with LPS resulted in an increase in the level of tyrosine phosphorylation of Btk (indicative of activation). The autokinase activity of Btk was also stimulated after LPS stimulation. In addition, a dominant negative form of Btk inhibited TLR4-mediated activation of a nuclear factor kappaB (NFkappaB)-dependent reporter gene in HEK293 cells as well as LPS-induced activation of NFkappaB in the astrocytoma cell line U373 and the monocytic cell line RAW264.7. Further investigation revealed that the Btk-specific inhibitor, LFM-A13, inhibited the activation of NFkappaB by LPS in THP-1 cells. Our findings implicate Btk as a Toll/interleukin-1 receptor domain-binding protein that is important for NFkappaB activation by TLR4.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [8] |
Bruton's tyrosine kinase (Btk) has recently been shown to participate in the induction of nuclear factor kappaB (NFkappaB)-dependent gene expression by the lipopolysaccharide (LPS) receptor Toll-like receptor-4 (TLR4). In this study we have examined the mechanism whereby Btk participates in this response. Treatment of the murine monocytic cell line Raw264.7 with LFM-A13, a specific Btk inhibitor, blocked LPS-induced NFkappaB-dependent reporter gene expression but not IkappaB alpha degradation. Transient transfection of HEK293 cells with Btk had no effect on NFkappaB-dependent reporter gene expression but strongly promoted transactivation of a reporter gene by a p65-Gal4 fusion protein. IkappaB alpha degradation activated by LPS was intact in macrophages from X-linked immunodeficiency (Xid) mice, which contain inactive Btk. Transfection of cells with a dominant negative form of Btk (BtkK430R) inhibited LPS-driven p65 mediated transactivation. Additionally LFM-A13 impaired phosphorylation of serine 536 on p65 induced by LPS in HEK293-TLR4 cells, and in Xid macrophages this response was impaired. This study therefore reveals a novel function for Btk. It is required for the signaling pathway activated by TLR4, which culminates in phosphorylation of p65 on serine 536 promoting transactivation by NFkappaB.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [9] |
IgA Fc receptors (FcαR) can mediate a variety of inflammatory responses. It has been demonstrated that the FcRγ subunit is critical in mediating signaling through FcαR. We show that aggregation of FcαR on U937 cells and blood neutrophils results in tyrosine phosphorylation of several intracellular proteins, including the FcR γ subunit, p72syk, and Bruton tyrosine kinase (Btk). Syk was found to be associated with FcαR and its phosphorylation was increased in phorbol myristate acetate (PMA)- and interferon-γ (IFN-γ)-treated U937 cells. In contrast, phosphorylation of Btk was only detected after cell treatment with PMA but not IFN-γ. These data indicate that signaling through FcαR γ2 involves at least two subfamilies of tyrosine kinases, syk and Btk. Our results also suggest that activation of tyrosine kinase pathways through FcαR depends on the activation state of the cell. This may be an important regulatory mechanism in IgA-mediated responses at inflammatory sites.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [10] |
Autoantibody production and immune complex deposition within the kidney promote renal disease in patients with lupus nephritis. Thus, therapeutics that inhibit these pathways may be efficacious in the treatment of systemic lupus erythematosus. Bruton's tyrosine kinase (BTK) is a critical signaling component of both BCR and FcR signaling. We sought to assess the efficacy of inhibiting BTK in the development of lupus-like disease, and in this article describe (R)-5-amino-1-(1-cyanopiperidin-3-yl)-3-(4-[2,4-difluorophenoxy]phenyl)-1H-pyrazole-4-carboxamide (PF-06250112), a novel highly selective and potent BTK inhibitor. We demonstrate in vitro that PF-06250112 inhibits both BCR-mediated signaling and proliferation, as well as FcR-mediated activation. To assess the therapeutic impact of BTK inhibition, we treated aged NZBxW_F1 mice with PF-06250112 and demonstrate that PF-06250112 significantly limits the spontaneous accumulation of splenic germinal center B cells and plasma cells. Correspondingly, anti-dsDNA and autoantibody levels were reduced in a dose-dependent manner. Moreover, administration of PF-06250112 prevented the development of proteinuria and improved glomerular pathology scores in all treatment groups. Strikingly, this therapeutic effect could occur with only a modest reduction observed in anti-dsDNA titers, implying a critical role for BTK signaling in disease pathogenesis beyond inhibition of autoantibody production. We subsequently demonstrate that PF-06250112 prevents proteinuria in an FcR-dependent, Ab-mediated model of glomerulonephritis. Importantly, these results highlight that BTK inhibition potently limits the development of glomerulonephritis by impacting both cell- and effector molecule-mediated pathways. These data provide support for evaluating the efficacy of BTK inhibition in systemic lupus erythematosus patients.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [11] |
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [12] |
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [13] |
The most problematic issue in clinical nephrology is the relentless and progressive increase in patients with ESRD (end-stage renal disease) worldwide. The impact of diabetic nephropathy on the increasing population with CKD (chronic kidney disease) and ESRD is enormous. Three major pathways showing abnormality of intracellular metabolism have been identified in the development of diabetic nephropathy: (i) the activation of polyol and PKC (protein kinase C) pathways; (ii) the formation of advanced glycation end-products; and (iii) intraglomerular hypertension induced by glomerular hyperfiltration. Upstream of these three major pathways, hyperglycaemia is the major driving force of the progression to ESRD from diabetic nephropathy. Downstream of the three pathways, microinflammation and subsequent extracellular matrix expansion are common pathways for the progression of diabetic nephropathy. In recent years, many researchers have been convinced that the inflammation pathways play central roles in the progression of diabetic nephropathy, and the identification of new inflammatory molecules may link to the development of new therapeutic strategies. Various molecules related to the inflammation pathways in diabetic nephropathy include transcription factors, pro-inflammatory cytokines, chemokines, adhesion molecules, Toll-like receptors, adipokines and nuclear receptors, which are candidates for the new molecular targets for the treatment of diabetic nephropathy. Understanding of these molecular pathways of inflammation would translate into the development of anti-inflammation therapeutic strategies.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [14] |
Bruton's tyrosine kinase (Btk) is encoded by the gene that when mutated causes the primary immunodeficiency disease X-linked agammaglobulinemia (XLA) in humans and X-linked immunodeficiency (Xid) in mice. Btk is a member of the Tec family of protein tyrosine kinases (PTKs) and plays a vital, but diverse, modulatory role in many cellular processes. Mutations affecting Btk block B-lymphocyte development. Btk is conserved among species, and in this review, we present the sequence of the full-length rat Btk and find it to be analogous to the mouse Btk sequence. We have also analyzed the wealth of information compiled in the mutation database for XLA (BTKbase), representing 554 unique molecular events in 823 families and demonstrate that only selected amino acids are sensitive to replacement (P < 0.001). Although genotype-phenotype correlations have not been established in XLA, based on these findings, we hypothesize that this relationship indeed exists. Using short interfering-RNA technology, we have previously generated active constructs downregulating Btk expression. However, application of recently established guidelines to enhance or decrease the activity was not successful, demonstrating the importance of the primary sequence. We also review the outcome of expression profiling, comparing B lymphocytes from XLA-, Xid-, and Btk-knockout (KO) donors to healthy controls. Finally, in spite of a few genes differing in expression between Xid- and Btk-KO mice, in vivo competition between cells expressing either mutation shows that there is no selective survival advantage of cells carrying one genetic defect over the other. We conclusively demonstrate that for the R28C-missense mutant (Xid), there is no biologically relevant residual activity or any dominant negative effect versus other proteins.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [15] |
Accumulating evidence has shown that macrophages play a vital role in development and pathogenesis of diabetic nephropathy (DN) by secreting inflammatory cytokines. Although Bruton's tyrosine kinases (Btk) is a biologically important molecule implicated in immune regulation, the role of Btk in high glucose (HG)-stimulated inflammatory response in macrophages and the mechanism involved need further investigation. In our study, we used bone marrow-derived macrophages (BMMs) to investigate the involvement of Btk on HG-induced inflammatory cytokines expression and to explore the underlying mechanisms. We found that high glucose induced phosphorylation of Btk, MAPKs and NF-κB, and the expression of downstream inflammation cytokines monocyte chemo-attractant protein-1 (MCP-1), tumor necrosis factor-alpha (TNF-α) and interleukin-1beta (IL-1β). Btk inhibitor (PCI-32765) not only down-regulated ERK1/2 phosphorylation and NF-κB activation, but also decreased the secretion of MCP-1, TNF-α and IL-1β in HG-treated BMMs. These results indicate that Btk plays an important role in HG-induced inflammatory cytokines expression and that PCI-32765 may be used as an immunoregulatory agent against hyperglycemia-induced inflammatory response in DN.Copyright © 2018. Published by Elsevier B.V.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [16] |
In several models of progressive glomerular disease, mesangial cell proliferation, phenotypic change and increased growth factor expression precede up-regulation of genes for extracellular matrix components (ECM) and mesangial expansion. To examine these events in diabetic nephropathy (DN) we conducted sequential studies of glomeruli in rats with streptozotocin induced DN. We found prominent mesangial cell proliferation at three days (4.34 +/- 2.24 PCNA + cells/glom vs. 1.6 +/- 0.74 in controls, P < 0.001) associated with increased alpha-actin expression. PDGF B-chain mRNA was slightly increased at day one, and PDGF B-chain immunostaining was slightly increased at days one and six. Staining for bFGF was significantly increased at three days (2.2 +/- 0.6 vs. 1.2 +/- 0.1 in controls, P < 0.01). There was also an early increase in platelets in glomeruli of diabetic animals, and platelet depletion significantly inhibited the early phase of proliferation. In addition to mesangial cell proliferation, a prominent glomerular macrophage infiltration began at day three and peaked at day 30 (3.94 +/- 1.47 vs. 2.08 +/- 1.13 in controls, P < 0.01). TGF-beta mRNA increased at days 14 and 30. Insulin treatment prevented mesangial cell proliferation, actin expression, and macrophage infiltration, and normalized TGF-beta expression at 14 and 30 days. These multiple cellular events preceded any detectable increases in glomerular gene expression or deposition of collagen I, IV or laminin.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [17] |
Diabetic kidney disease (DKD) is the leading cause of kidney failure worldwide and the single strongest predictor of mortality in patients with diabetes. DKD is a prototypical disease of gene and environmental interactions. Tight glucose control significantly decreases DKD incidence, indicating that hyperglycemia-induced metabolic alterations, including changes in energy utilization and mitochondrial dysfunction, play critical roles in disease initiation. Blood pressure control, especially with medications that inhibit the angiotensin system, is the only effective way to slow disease progression. While DKD is considered a microvascular complication of diabetes, growing evidence indicates that podocyte loss and epithelial dysfunction play important roles. Inflammation, cell hypertrophy, and dedifferentiation by the activation of classic pathways of regeneration further contribute to disease progression. Concerted clinical and basic research efforts will be needed to understand DKD pathogenesis and to identify novel drug targets.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [18] |
张洋, 马坤岭, 刘晶, 等. 慢性炎性反应对糖尿病肾病的促进作用[J]. 中华肾脏病杂志, 2013, 29(9): 681-686. DOI:10.3760/cma.j.issn.1001-7097.2013.09.008.
目的 通过制备糖尿病肾病微炎性反应动物模型,探讨慢性炎性反应在糖尿病肾病进展中的作用及意义。 方法 选取8周龄雄性db/db小鼠及对照组db/m小鼠,分别按随机数字表法分为db/db、db/db+酪蛋白组和db/m、db/m+酪蛋白组,每组均为8只。db/m+酪蛋白组及db/db+酪蛋白组隔日给予背部皮下注射10%酪蛋白溶液0.5 ml以刺激产生慢性、持续性、微炎性反应;db/m组及db/db组隔日给予背部皮下注射蒸馏水0.5 ml。每周称体质量、收集24 h尿液、检测24 h尿蛋白量,8周后处死,收集血清标本、留取肾组织,检测血清淀粉样蛋白A(SAA)、肿瘤坏死因子α(TNF-α)浓度,病理染色及电镜检查观察肾小球病理改变,免疫组化及Western印迹法观察肾脏炎性因子及足细胞特异性标志蛋白的表达情况,并评估微炎性反应模型的建立在糖尿病肾病研究中的作用及意义。 结果 db/m+酪蛋白组及db/db+酪蛋白组血清炎性因子SAA[(13.83±0.29) mg/L 比(1.52±0.19) mg/L,P<0.05;(13.84±0.28) mg/L比(1.67±0.58) mg/L,P<0.05]及TNF-α[(14.23±1.42) ng/L比(10.70±1.38) ng/L,P<0.05;(14.54±1.91) ng/L 比(10.88±1.22) ng/L,P<0.05]水平均显著高于其对照组,且肾组织中单核细胞趋化蛋白1(MCP-1)、TNF-α蛋白表达亦高于其对照组;db/db+酪蛋白组小鼠尿蛋白量、肾小球病理改变、足突结构改变及数量减少程度与db/db组相比明显加重,但db/m+酪蛋白组与db/m组间无明显差别。 结论 本研究通过构建糖尿病肾病微炎性反应模型证实,持续存在的慢性微炎性反应在加速糖尿病肾病进展中扮演着重要作用。
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [19] |
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [20] |
Chronic kidney disease (CKD) has become a major health problem worldwide. This review describes the role of macrophages in CKD and highlights the importance of anti-inflammatory M2 macrophage activation in both renal fibrosis and wound healing processes. Furthermore, the mechanisms by which M2 macrophages induce renal repair and regeneration are still under debate and currently demand more attention. The M1/M2 macrophage balance is related to the renal microenvironment and could influence CKD progression. In fact, an inflammatory renal environment and M2 plasticity can be the major hurdles to establishing macrophage cell-based therapies in CKD. M2 macrophage cell-based therapy is promising if the M2 phenotype remains stable and is 'fixed' by manipulation. However, a greater understanding of phenotype polarization is still required. Moreover, better strategies and targets to induce reparative macrophages should guide future investigations in order to abate kidney diseases.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [21] |
Cytokines act as pleiotropic polypeptides regulating inflammatory and immune responses through actions on cells. They provide important signals in the pathophysiology of a range of diseases, including diabetes mellitus. Chronic low-grade inflammation and activation of the innate immune system are closely involved in the pathogenesis of diabetes and its microvascular complications. Inflammatory cytokines, mainly IL-1, IL-6, and IL-18, as well as TNF-alpha, are involved in the development and progression of diabetic nephropathy. In this context, cytokine genetics is of special interest to combinatorial polymorphisms among cytokine genes, their functional variations, and general susceptibility to diabetic nephropathy. Finally, the recognition of these molecules as significant pathogenic mediators in diabetic nephropathy leaves open the possibility of new potential therapeutic targets.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [22] |
The NF-κB family of transcription factors has an essential role in inflammation and innate immunity. Furthermore, NF-κB is increasingly recognized as a crucial player in many steps of cancer initiation and progression. During these latter processes NF-κB cooperates with multiple other signaling molecules and pathways. Prominent nodes of crosstalk are mediated by other transcription factors such as STAT3 and p53 or the ETS related gene ERG. These transcription factors either directly interact with NF-κB subunits or affect NF-κB target genes. Crosstalk can also occur through different kinases, such as GSK3-β, p38, or PI3K, which modulate NF-κB transcriptional activity or affect upstream signaling pathways. Other classes of molecules that act as nodes of crosstalk are reactive oxygen species and miRNAs. In this review, we provide an overview of the most relevant modes of crosstalk and cooperativity between NF-κB and other signaling molecules during inflammation and cancer.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| [23] |
Thiazolidinedione (TZD), a ligand for peroxisome proliferator-activated receptor-γ (PPAR-γ), exerts anti-inflammatory effects independently of the insulin-sensitizing effect. In the present study, we tested the hypothesis that TZD prevents the progression of diabetic nephropathy by modulating the inflammatory process. Five-week-old Sprague-Dawley rats were divided into three groups: 1) nondiabetic control rats (non-DM), 2) diabetic rats (DM), and 3) diabetic rats treated with pioglitazone (DM+pio). Diabetes was induced by injection with streptozotocin (STZ). The DM+pio group received 0.0002% pioglitazone mixed in chow for 8 wk after induction of diabetes. Blood glucose and HbA1c were elevated in diabetic rats but did not change by treatment with pioglitazone. Pioglitazone reduced urinary albumin excretion and glomerular hypertrophy, suppressed the expression of transforming growth factor (TGF)-β, type IV collagen, and ICAM-1, and infiltration of macrophages in the kidneys of diabetic rats. Furthermore, renal NF-κB activity was increased in diabetic rats and reduced by pioglitazone. PPAR-γ was expressed in glomerular endothelial cells in the diabetic kidney and in cultured glomerular endothelial cells. High-glucose conditions increased the expression of ICAM-1 and the activation of NF-κB in cultured glomerular endothelial cells. These changes were reduced by pioglitazone, ciglitazone, and pyrrolidine dithiocarbamate, an inhibitor of NF-κB. However, pioglitazone did not show the changes in the presence of PPAR-γ antagonist GW9662. Our results suggest that the preventive effects of pioglitazone may be mediated by its anti-inflammatory actions, including inhibition of NF-κB activation, ICAM-1 expression, and macrophage infiltration in the diabetic kidney.
{{custom_citation.content}}
{{custom_citation.annotation}}
|
| {{custom_ref.label}} |
{{custom_citation.content}}
{{custom_citation.annotation}}
|
PDF(27228 KB)
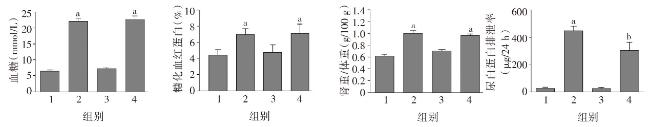
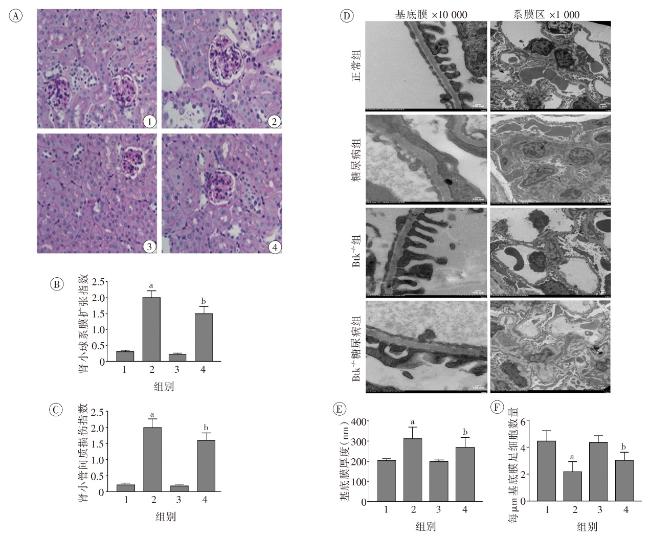
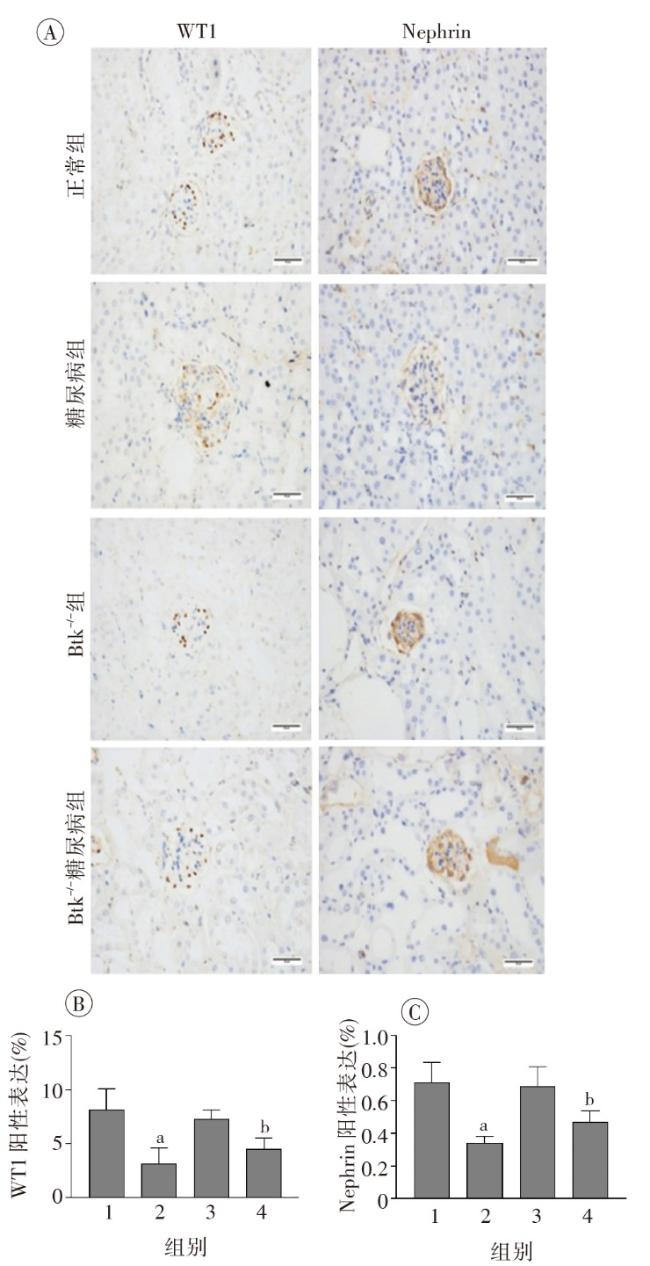
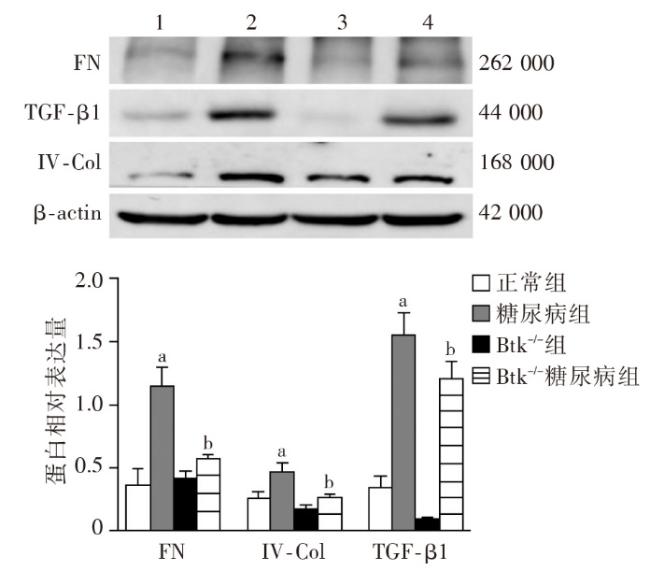
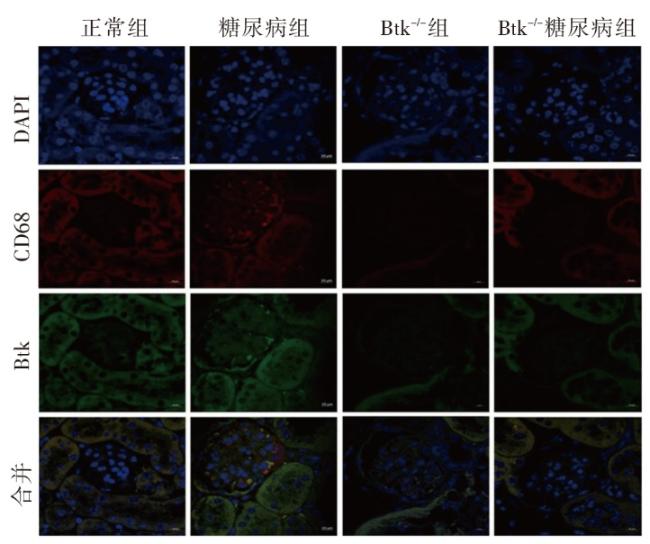
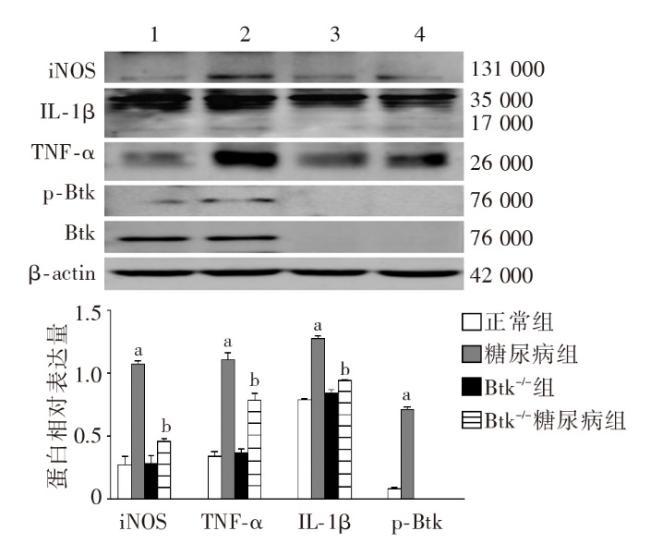
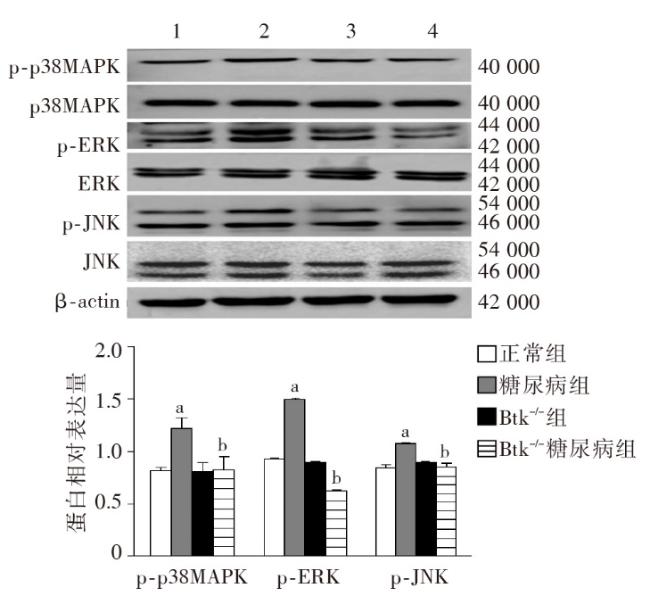
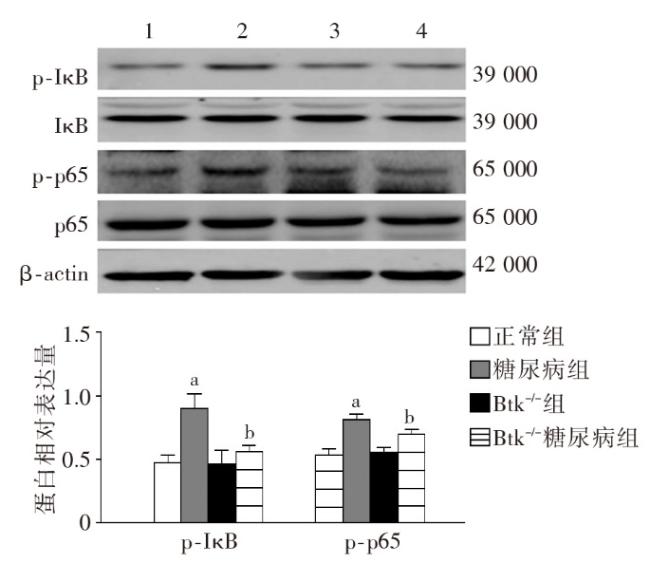
 图1 各组小鼠一般代谢指标比较图2 各组小鼠肾脏组织病理学改变图3 各组小鼠肾脏足细胞标志物WT1和Nephrin的表达图4 各组小鼠肾脏组织蛋白FN、IV-Col和TGF-β1的表达变化(Western印迹)图5 各组小鼠肾脏巨噬细胞浸润和活化指标CD68和Btk的表达(激光共聚焦 ×400)图6 各组小鼠肾组织炎性因子IL-1β、TNF-α 和MCP-1 mRNA的表达(实时荧光定量PCR)图7 各组小鼠肾脏组织iNOS、IL-1β、TNF-α和p-Btk蛋白的表达(Western印迹)图8 各组小鼠肾脏p-p38MAPK、p-ERK和p-JNK蛋白表达(Western印迹)图9 各组小鼠肾脏中p-IκB和p-p65蛋白的表达(Western印迹)
图1 各组小鼠一般代谢指标比较图2 各组小鼠肾脏组织病理学改变图3 各组小鼠肾脏足细胞标志物WT1和Nephrin的表达图4 各组小鼠肾脏组织蛋白FN、IV-Col和TGF-β1的表达变化(Western印迹)图5 各组小鼠肾脏巨噬细胞浸润和活化指标CD68和Btk的表达(激光共聚焦 ×400)图6 各组小鼠肾组织炎性因子IL-1β、TNF-α 和MCP-1 mRNA的表达(实时荧光定量PCR)图7 各组小鼠肾脏组织iNOS、IL-1β、TNF-α和p-Btk蛋白的表达(Western印迹)图8 各组小鼠肾脏p-p38MAPK、p-ERK和p-JNK蛋白表达(Western印迹)图9 各组小鼠肾脏中p-IκB和p-p65蛋白的表达(Western印迹)/
| 〈 |
|
〉 |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}